1. My Apollo window has frozen. What do I do?
Try reloading your browser window.
- For Chrome: Shift Command R
- For Firefox: Ctrl+R
- For Safari: shift+reload
2. Some data tracks are very slow to load.
The way JBrowse works within Apollo, the first time your load up a track it might take a while. But once loaded, any successive loads will be quicker.
3. I used to see tracks and now they are gone.
Close the track and open it again. Yes, this is the standard 'turn it off and turn it on again' solution.
4. What is the meaning of the lower case letters in the nucleotide sequence?
These indicate regions of low complexity. They have no impact/implication with respect to annotation.

5. Is it possible to know which organism is the source for a particular protein alignment (in the Protein Alignment track)?
By right clicking on the alignment, and then selecting 'View details.' You'll see the GenBank accession in the id section and can search for that at NCBI.

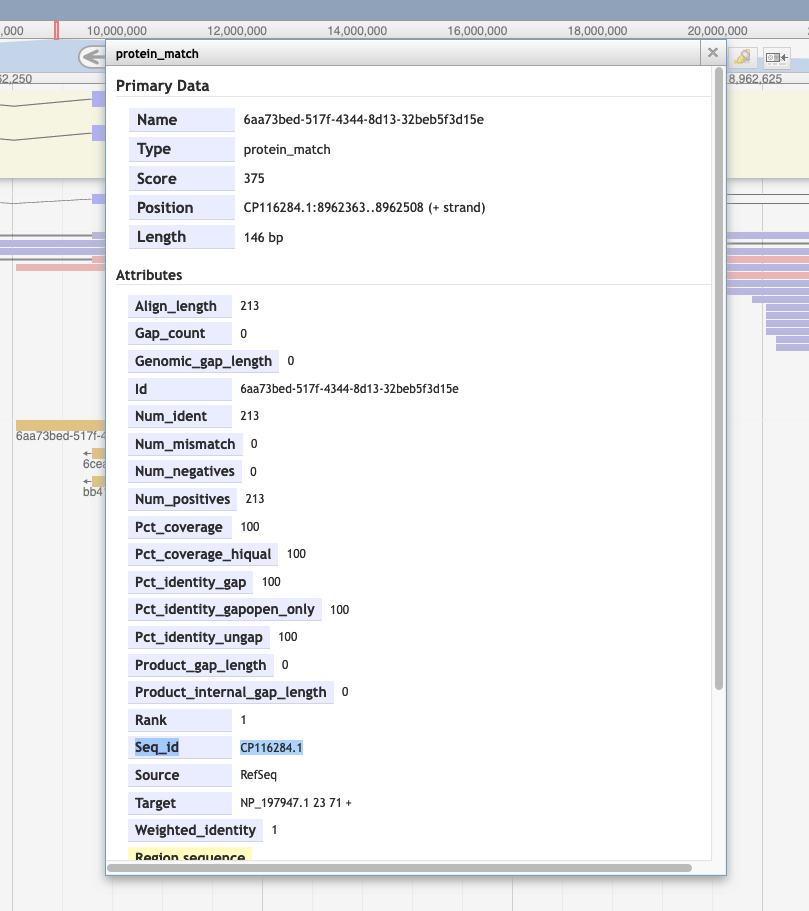
6. Where is the track metadata?




TRACK NAME | Description of data |
Col-CC_Genomic_Annotations_Data | Result of NCBI Eukaryotic Annotation Pipeline |
AT-Col-CC-Liftoff-from-TAIR10.1 | v11 models mapped to v12 reference using Liftoff |
| TranscriptomeReconstructoR models | Method: (1) Assembled expression evidence: ONT-DRS (ERR3764345 - ERR3764351), CAGE-Seq (SRR10045003 - SRR10045005), PAT-Seq (SRR7160296, SRR7160297, SRR7160299), plaNET-Seq (SRR9117170 - SRR9117173) (2) Aligned all the datasets to Col-CC genome (3) Built TRR based annotation using alignment (bam) files Output:
|
Gnomon Models | One of the outputs of the annotation pipeline. These are a superset of the final set of annotated models. "Gnomon annotation of the genomic sequence. Sequence identifiers are provided as accession.version for the genomic sequences and Gnomon identifiers for the Gnomon models:gene.XXX for genes, GNOMON.XXX.m for transcripts and GNOMON.XXX.p for proteins. These identifiers are NOT universally unique. They are unique per annotation release only." (from NCBI documentation) |
| Coverage Tracks, RNAseq and Long Read | |
| RNAseq capped_merged_filtered coverage | Coverage track version of the RNAseq capped_merged_filtered reads. |
| Long Read Alignments coverage, plus strand | Coverage track of the long reads, plus strand ONLY. |
| Long Read Alignments coverage, minus strand | Coverage track of the long reads, minus strand ONLY. |
| Merged, Capped, and Filtered RNAseq Reads | |
| RNAseq capped_merged_filtered reads | RNAseq reads from the 62 RNAseq experiments after filtering for overly long (>5Kbp) inserts, and successive capping and merging to try to reduce the overabundance of highly expressed genes. File was then filtered to remove overly long (>12Kbp) introns. |
| Protein Evidence | |
| PFAM domains | Results from an INTERPROSCAN run on the proteins from the V12 prediction to get the PFAM domain information, converted to absolute position on the Col-CC assembly. |
| PFAM domains - Liftoff | Results from an INTERPROSCAN run on the proteins from the Araport11 release to get the PFAM domain information, converted to absolute position on the Col-CC assembly (using the Liftoff file that converted Araport11 coordinates to Col-CC coordinates). |
| PANTHER families | Results from an INTERPROSCAN run on the proteins from the V12 prediction to get the PANTHER family information, converted to absolute position on the Col-CC assembly. |
| PANTHER families - Liftoff | Results from an INTERPROSCAN run on the proteins from the Araport11 release to get the PANTHER family information, converted to absolute position on the Col-CC assembly (using the Liftoff file that converted Araport11 coordinates to Col-CC coordinates). |
| Protein alignments chained | Alignments of Arabidopsis thaliana and other Brassicaceae proteins, including Araport 11 annotated proteins, to the genomic sequence(s). These alignments may have been used as evidence for gene prediction by the NCBI annotation pipeline. Pieces of the same protein have been connected together for easier visualization. |
| TRR CAGE PAT plaNET | |
| CAGE forward | TranscriptomeReconstructoR models CAGE evidence, forward strand (evidence supporting the start site of transcription) |
| CAGE reverse | TranscriptomeReconstructoR models CAGE evidence, reverse strand (evidence supporting the start site of transcription) |
| PAT forward | TranscriptomeReconstructoR models PAT evidence, forward strand (evidence supporting the end site of transcription) |
| PAT reverse | TranscriptomeReconstructoR models PAT evidence, reverse strand (evidence supporting the end site of transcription) |
| plaNET forward | TranscriptomeReconstructoR models plaNET evidence, forward strand (evidence supporting transcription of mRNAs and lncRNAs) |
| plaNET reverse | TranscriptomeReconstructoR models plaNET evidence, reverse strand (evidence supporting transcription of mRNAs and lncRNAs) |
| Transcript Evidence | |
| Known Reference Sequences | "Alignments of the annotated Known RefSeq transcripts (identified with accessions prefixed with NM_ and NR_) to the genome." (from NCBI documentation) These were NOT used in generating the Col-CC annotation. They are alignments of the annotated transcripts to the genome and can provide additional insight into the predicted gene structures independent of the prediction. |
| Model Reference Sequences | "Alignments of the annotated Model RefSeq transcripts (identified with accessions prefixed with XM_ and XR_) to the genome." (from NCBI documentation) These were NOT used in generating the Col-CC annotation. They are alignments of the annotated transcripts to the genome and can provide additional insight into the predicted gene structures independent of the prediction. |
| Col-CC Same Species Combined | Alignments of same-species cDNAs, ESTs and TSAs to the genomic sequence(s). cDNAs and ESTs alignments (not TSAs) may have used as evidence for gene prediction by the NCBI annotation pipeline. The TSA alignment track is a subset of the Col-CC Same Species track. Pieces of the same transcript have been connected together for easier visualization. |
| TSA alignment | Alignments of transcripts assembled from RNA-Seq reads, and submitted to GenBank (see accessions DAHAIV01, GGJX01, GJRK01 and GKIF01). These were not used as evidence for gene prediction by the NCBI annotation pipeline. |
RNA seq tracks from various plant parts and growth stages/conditions of those parts | Name is based on the GenBank record, for example, SRR1019221. You can link to that record using this base URL for more information on the experiment: |
| Long Read alignments | Alignments of individual IsoSeq reads in SRA. These alignments may have been used as evidence for gene prediction by the NCBI annotation pipeline. Right clicking on the read itself will allow you to ‘View Details’ and see the ID of the SRA entry for the experiment. Using the id (e.g., SRR11031292), you can go to the full GenBank record for the experiment. https://www.ncbi.nlm.nih.gov/sra/?term=SRR11031292. |
7. What does the warning symbol mean?
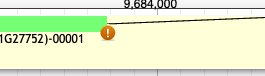
This symbol means that the location and sequence of the splice site needs to be investigated and verified because it does not conform to the GT-AG rule. Other possibilities include GC-AG and AT-AC.
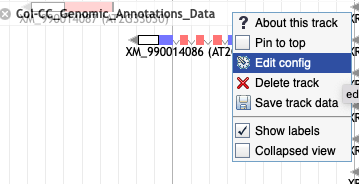
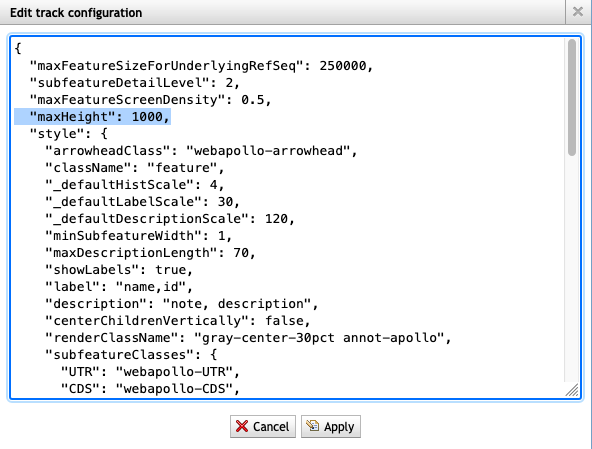
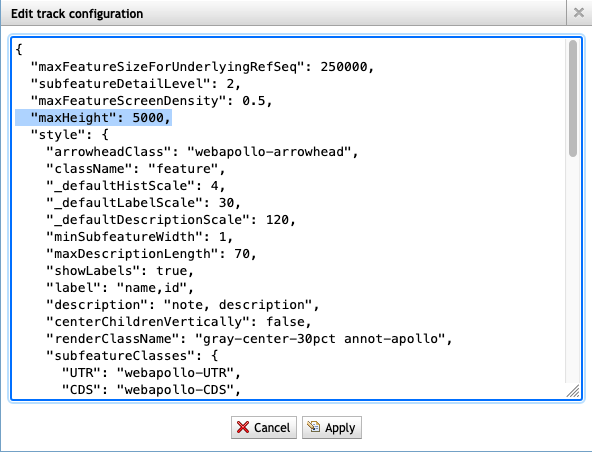
8. The track I've viewing says 'Max height reached' but I want to see more rows. How do I do that?
- Click on Edit config.
- Scroll to the top of the config, find the ‘maxHeight’ setting.
- Change the value to ‘5000’
- Click on ‘Apply’
- See more rows!
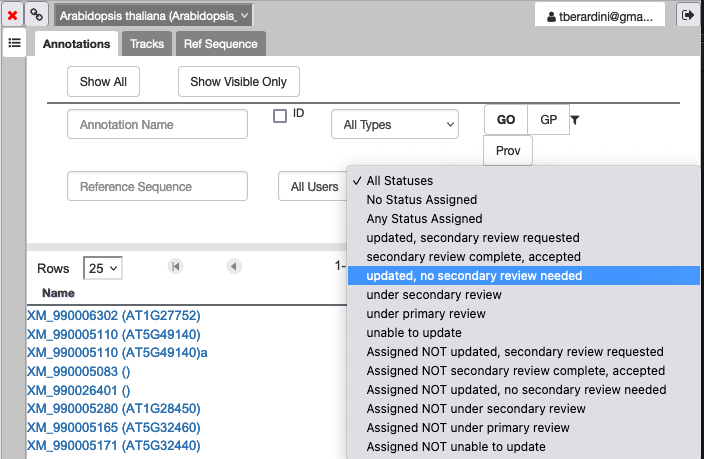
9. How do I see genes that need secondary review?
In the Annotations tab of your right hand panel, click on the dropdown for "All Statuses" and select the status you want to filter the annotations to review.
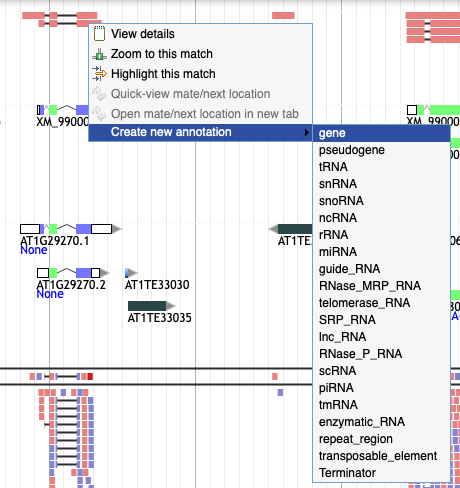
10. I want to drag an element from an evidence track to the user-created annotation track but I can't!
Right click on the element and select 'Create new annotation' from the menu. Pick the type of element you want to create and it will appear in the yellow track.
11. Comment/Renaming-related questions
11.1. Where do I add my comment/rename?
- If you have already added comments at the gene level, these will apply to all mRNAs under that gene.
- In cases where there is only one mRNA/gene, the comment/renaming (DELETE) on the mRNA transitively applies to the gene.
- If there is more than one mRNA/gene and you want to record different actions for individual mRNAs, please put the specific comments (or DELETE) on the relevant mRNAs only.
- If there is more than one mRNA/gene and you want to record the same action for ALL mRNAs/the whole gene, please put the specific comments (or DELETE) on any one of the mRNAs.
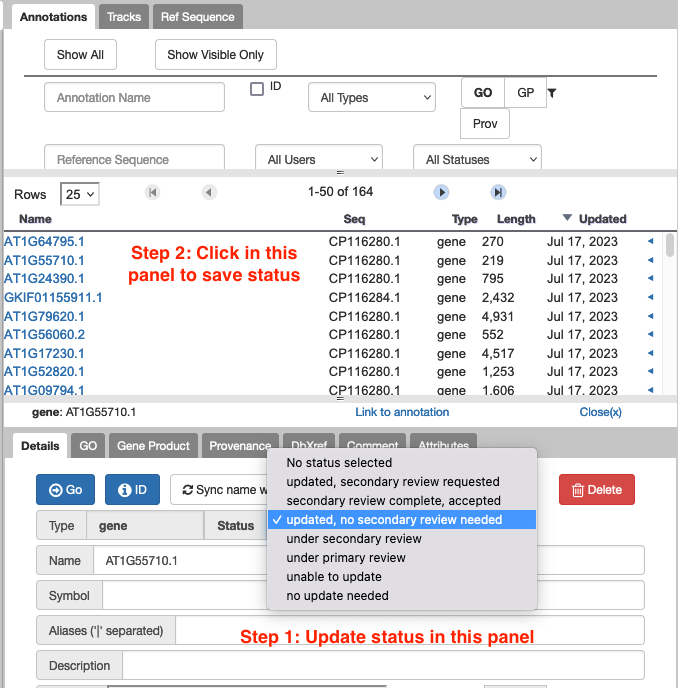
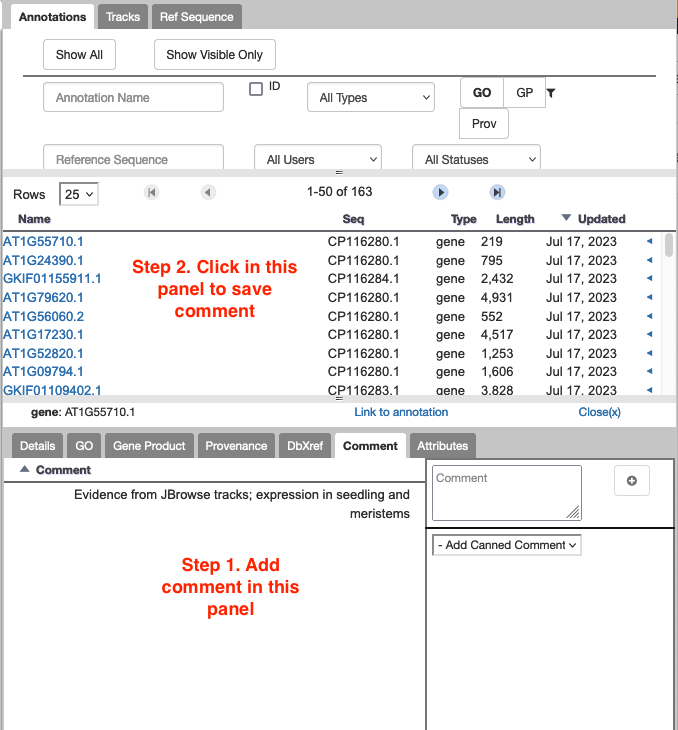
11.2. The comment or status I typed in hasn't saved. How do I make sure it saves?
For saving comments and gene status, make sure you click outside of the panel where you created the comment to ‘make it stick’.
11.3. The deleted gene I'm reviewing needs to stay deleted. How do I mark this in Apollo?
- 1. create a gene model of the gene that should stay deleted by dragging into the user-created annotations
- 2. rename so that the name ends with ‘DELETE’
- 3. set status (either ‘updated, secondary review requested’ OR ‘updated, no secondary review needed’)
- (see Q10 for what to do if there are different actions for different mRNAs of the same gene)
11.4. Check the names of the genes in the user-created annotations track to maintain AGI history.
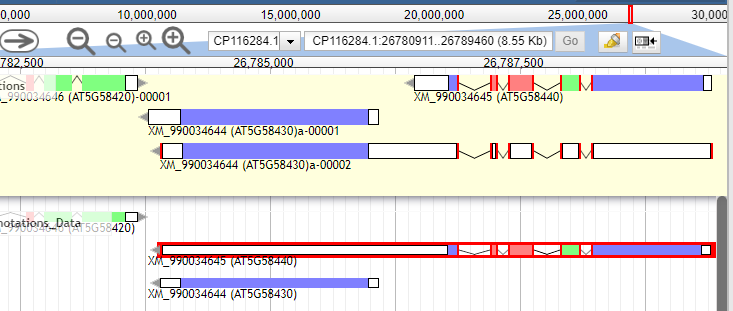
When dragging an annotation from the Col-CC to the user-created annotations, if the gene range is overlapping with the gene on the left, the gene ID will be replaced by the gene on the left. Just want to bring awareness to this small bug so that we don't mistakenly assign gene IDs to the user-created annotation track.
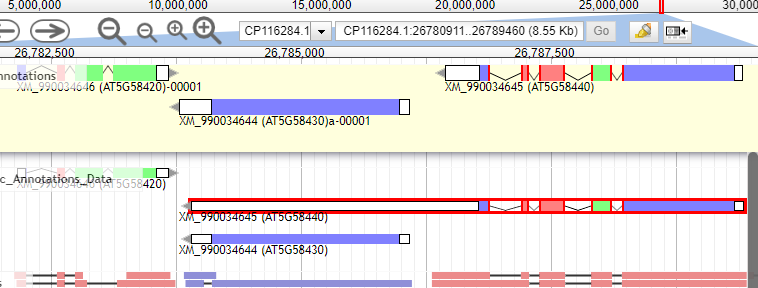
In this example, the AT5G58440 has a mistakenly annotated 3'UTR, that was merged with AT5G58430. When I dragged AT5G58440 from Col-CC track to user-created annotation track, the gene ID was replaced by AT5G58430. So the gene ID needs to be curated in this case. (Thanks, Xiaohui Li, for reporting.)
Before: (Dragged longer gene model has AGI = AT5G58430)
After: (Gene model to the 3' end after editing was renamed to be AGI = AT5G58440)
12. What do those clear/all white gene models mean? Should we keep them?

These gene models exist because the transcripts are made BUT they have been predicted to be targeted for Nonsense-Mediated Decay (NMD). No protein product is predicted to be made even though there may be a recognizable open reading frame. The gene models should be retained because they may have regulatory or other functions as RNAs.
NMD review: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7854845/
In the example above, the three all white gene models have subtle differences that can be seen when zooming in closer.
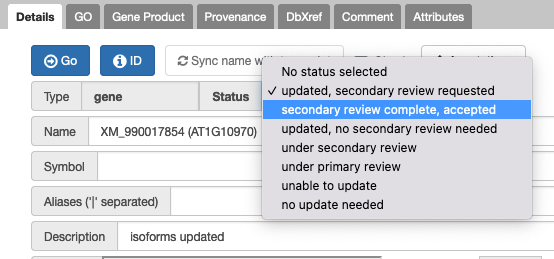
13. How does secondary review work?
- Go to Annotations tab in Apollo.
- Select status = ‘updated, secondary review requested’ from the drop down.

- See list of results (620 as of 10/17/23)
.
- Click on one of the results. (if it’s one that was originally yours, skip!)
- Click on the blue GO button.
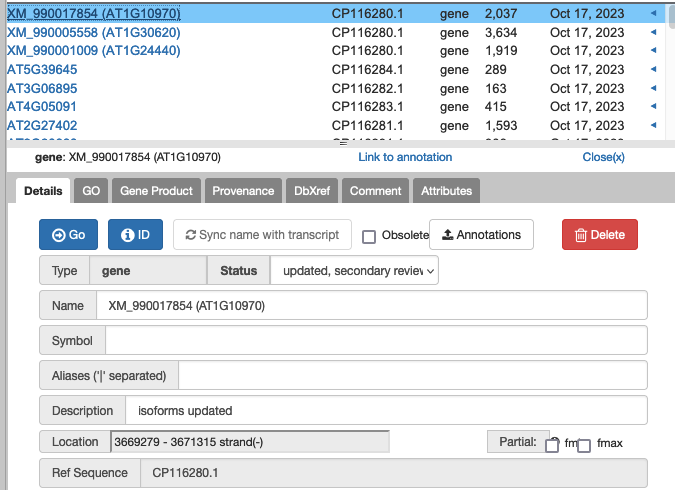
- The genome view will change to that location.
- Set the status to ‘under secondary review’ so no one else nabs this.
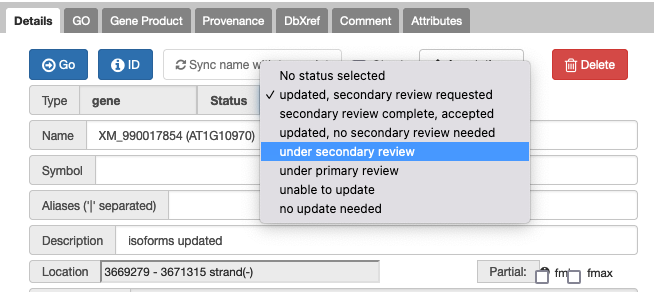
- TO FINISH: Review, edit, comment to track what you did/if you agree and finally change status to ‘secondary review completed, accepted’. (or select 'for discussion' if you can’t decide)
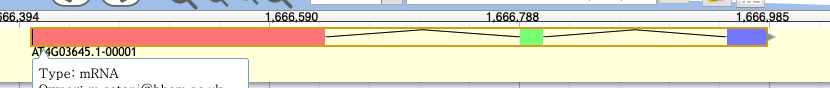
14. Apollo automatically changed the ORF when I dragged the gene model to the user created annotations band. How do I fix it?
Left click to highlight the whole gene model (in an intron) in the user-created annotations track.
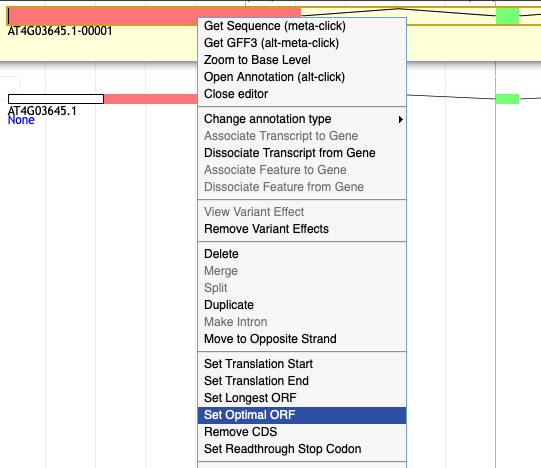
Option A. Right click and select 'Set Optimal ORF'. If that restores the v11 ORF, you are done.
Option B. Right click and select 'Zoom to Base Level'.
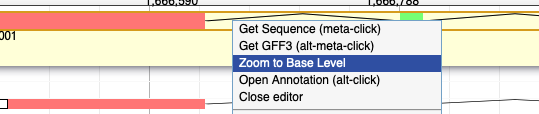
Find the ATG where the v11 gene model starts translation in the user created annotations version. Hover the cursor on the A.
Right click and select Set Translation Start. The v11 ORF should be restored.
