- What is a gene family in PhyloGenes?
- How is the boundary of a gene family defined?
- What is the procedure you used in building a gene family and constructing its gene tree?
- How are PANTHER gene trees constructed?
- How is branch length calculated?
- Why aren’t there any bootstrap or other support values for the tree topology?
- What is a subfamily?
- How are gene families named? Why are some families not named?
- How are subfamilies named. Why are some subfamilies not named?
- What is horizontal gene transfer? How is it detected in PANTHER gene trees?
- Are polyploid organisms represented in gene families?
- How does tree pruning work? Does pruning change the tree topology?
My gene is not found in PhyloGenes. Why?
What is a gene family in PhyloGenes?
Gene families in PhyloGenes are pruned versions <link to FAQ section> of PANTHER gene families (pantherdb.org, PMID:30407594). They contain only genes from selected plant genomes and 10 non-plant model organisms <link to list>. Genes from other genomes in the PANTHER build have been removed (pruned) from the PANTHER gene families and gene trees.
A PANTHER gene family contains genes that are related to each other by descent from a common ancestor, as established by statistical sequence similarity, and whose protein sequences can be aligned reliably into a multiple sequence alignment. The UniProt Reference Proteomes (https://www.ebi.ac.uk/reference_proteomes) used in PANTHER family construction contain one representative protein sequence per gene. A gene family is represented as a phylogenetic gene tree that shows how the family evolved by the processes of speciation, gene duplication, and horizontal transfer.
How is the boundary of a gene family defined?
In PANTHER, gene families are defined as clusters of related protein sequences (each protein sequence represents a distinct gene) for which a good multiple sequence alignment can be made (PubMed:23193289, PubMed:26578592). The basic requirements for a family are: (1) the family contains at least five sequences and includes more than one organism, and (2) the family has a sequence alignment of adequate quality to support phylogenetic inference. An alignment must have at least 30 sites aligned across 75% or more of the family members, and the derived Hidden Markov Model (HMM) must be able to recognize, with statistical significance, the sequences used to train it.
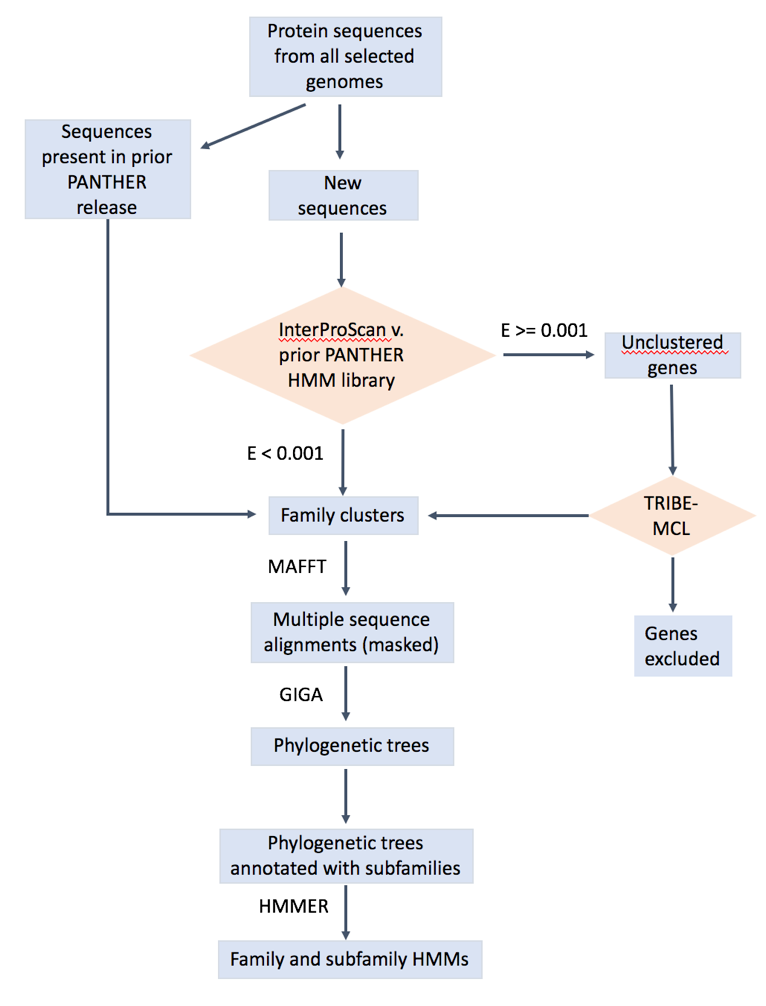
What is the procedure you used in building a gene family and constructing its gene tree?
The overall workflow is shown below. The details can be found in: https://www.ncbi.nlm.nih.gov/pubmed/23193289 (https://doi.org/10.1093/nar/gks1118), https://www.ncbi.nlm.nih.gov/pubmed/26578592, https://www.ncbi.nlm.nih.gov/pubmed/27899595