- What is a gene family in PhyloGenes?
- How is the boundary of a gene family defined?
- What is the procedure you used in building a gene family and constructing its gene tree?
- How are PANTHER gene trees constructed?
- How is branch length calculated?
- Why aren’t there any bootstrap or other support values for the tree topology?
- What is a subfamily?
- How are gene families named? Why are some families not named?
- How are subfamilies named. Why are some subfamilies not named?
- What is horizontal gene transfer? How is it detected in PANTHER gene trees?
- Are polyploid organisms represented in gene families?
- How does tree pruning work? Does pruning change the tree topology?
My gene is not found in PhyloGenes. Why?
What is a gene family in PhyloGenes?
Gene families in PhyloGenes are pruned versions <link to FAQ section> of PANTHER gene families (pantherdb.org, PMID:30407594). They contain only genes from selected plant genomes and 10 non-plant model organisms <link to list>. Genes from other genomes in the PANTHER build have been removed (pruned) from the PANTHER gene families and gene trees.
A PANTHER gene family contains genes that are related to each other by descent from a common ancestor, as established by statistical sequence similarity, and whose protein sequences can be aligned reliably into a multiple sequence alignment. The UniProt Reference Proteomes (https://www.ebi.ac.uk/reference_proteomes) used in PANTHER family construction contain one representative protein sequence per gene. A gene family is represented as a phylogenetic gene tree that shows how the family evolved by the processes of speciation, gene duplication, and horizontal transfer.
How is the boundary of a gene family defined?
In PANTHER, gene families are defined as clusters of related protein sequences (each protein sequence represents a distinct gene) for which a good multiple sequence alignment can be made (PubMed:23193289, PubMed:26578592). The basic requirements for a family are: (1) the family contains at least five sequences and includes more than one organism, and (2) the family has a sequence alignment of adequate quality to support phylogenetic inference. An alignment must have at least 30 sites aligned across 75% or more of the family members, and the derived Hidden Markov Model (HMM) must be able to recognize, with statistical significance, the sequences used to train it.
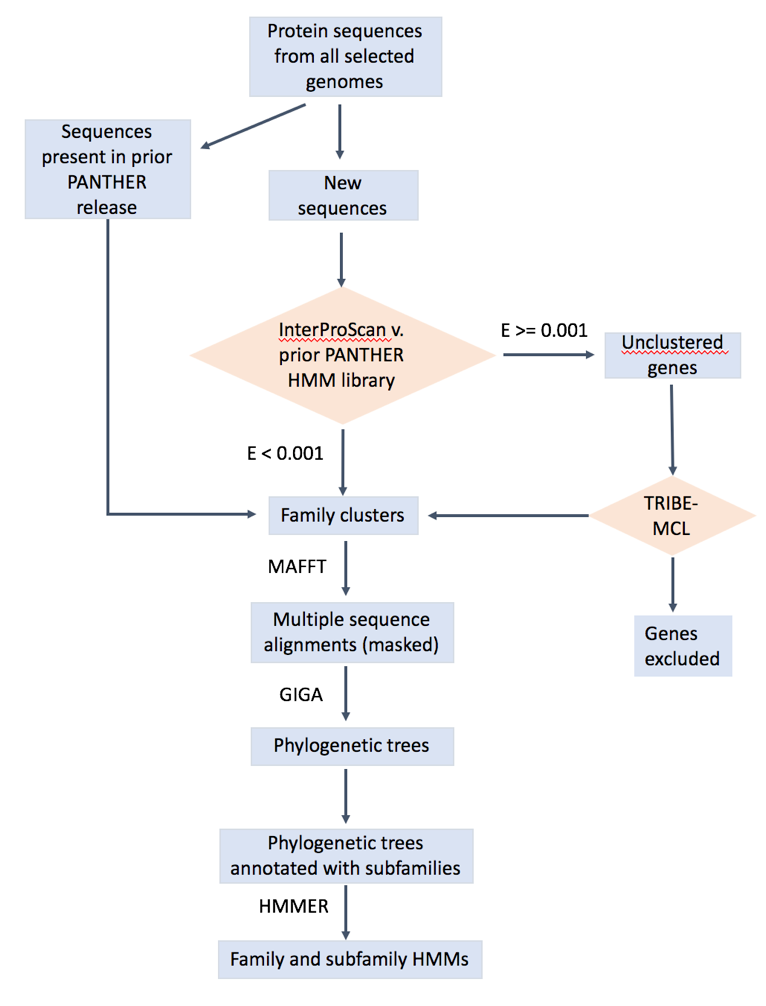
What is the procedure you used in building a gene family and constructing its gene tree?
The overall workflow is shown below. The details can be found in: https://www.ncbi.nlm.nih.gov/pubmed/23193289 (https://doi.org/10.1093/nar/gks1118), https://www.ncbi.nlm.nih.gov/pubmed/26578592, https://www.ncbi.nlm.nih.gov/pubmed/27899595
How is a PANTHER gene tree constructed?
A PANTHER gene tree is composed of orthologous subtrees (containing protein sequences related by speciation events), joined together by gene duplication (duplication within the same genome) or horizontal transfer (insertion from another genome). PANTHER trees were constructed by using the GIGA algorithm (https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-11-312v). The algorithm builds a tree from leaf nodes to the root, using a pairwise sequence distance matrix, a known species tree (based on NCBI taxonomy), and a set of rules to establish the tree topology. Sequence distance is calculated as the fraction of sequence differences between two sequences at selected homologous sites. The homologous sites were selected from multiple sequence alignment of all genes in the family. GIGA iteratively joins together subtrees of sequences, beginning with the two sequences that are closest according to the pairwise sequence distance matrix. The topology of the joined subtree after each iteration is not simply an agglomeration of the constituent subtrees. Rules are used to "rearrange" the joined subtree at each iteration. For example, if a subtree contains only speciation events, the topology is determined by the known species tree. Copying events such as duplication or horizontal transfer are placed within a tree with the most parsimonious solution to minimize gene deletions. The full description of the GIGA algorithm can be found in this paper (https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-11-312).
How is branch length calculated?
First, the ancestral sequence is inferred for each non-leaf node using a local, parsimony-like algorithm that reconstructs each node using only its descendants and closest outgroup. If over half of the descendant nodes align the same amino acid at a given site, it is inferred to be the most likely ancestral amino acid. If the descendants disagree, and the outgroup agrees with one of them, the outgroup amino acid is inferred to be the most likely ancestral amino acid. Otherwise, the ancestral amino acid is considered to be unknown ('X'). Next, the branch length between a parent node and a child node was calculated as the fraction of sequence differences between them. The Jukes-Cantor correction is applied to this value. More details can be found here (https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-11-312).
Why aren’t there any bootstrap or other support values for the tree topology?
Although the GIGA algorithm builds trees using a distance matrix, the topology of a tree is less influenced by sequences when compared to other distance or character-based algorithms such as neighbor joining or maximum likelihood. The reason is that GIGA imposes strict rules in determining tree topology. It uses a pre-established species tree in sorting out speciation nodes. GIGA uses maximum parsimony in placing duplication nodes. Therefore, the topology of a tree is very robust. For example, the rule of using species tree is illustrated below:

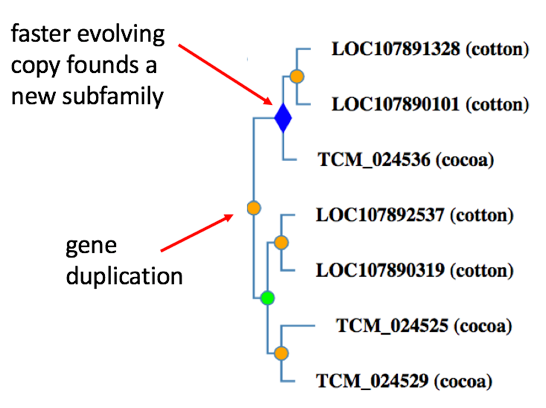
What is a subfamily?
Subfamilies within each family are groups of genes that share a particularly high degree of protein sequence similarity due to limited divergence from their common ancestor [https://www.ncbi.nlm.nih.gov/pubmed/26578592]. Subfamilies are, in general, closely-related orthologs. In the PANTHER tree building process, a new subfamily is created within a family after every gene duplication event, or horizontal transfer event. After horizontal transfer, the transferred copy becomes the founder of a new subfamily; the vertically inherited copy remains in the original subfamily. After gene duplication, the copy that changes faster in sequence immediately following the duplication becomes the founder of a new subfamily; the slower-evolving copy remains in the same subfamily. There are two exceptions to this rule: (1) because of the high frequency of gene duplication prior to the vertebrate common ancestor, each vertebrate copy following a gene duplication event founds a new subfamily, and (2) duplicated genes do not found a subfamily if they did not lead to orthologs in at least two extant species.